
VALIDATION
In a recent publication,we demonstrated that HOMINGS was largely equivalent to the tree-based approach implemented in QIIME. Community composition profiles of many clinical samples and mock communities were congruent between the two analysis approaches. Alpha and beta diversity of clinical samples were also very similar in comparing the 2 approaches.
As an example, in analyses of 10 mock communities each containing about 10 to 15 taxa, there was good correlation between QIIME and HOMINGS. Each method detected the appropriate taxon as shown in the figure below
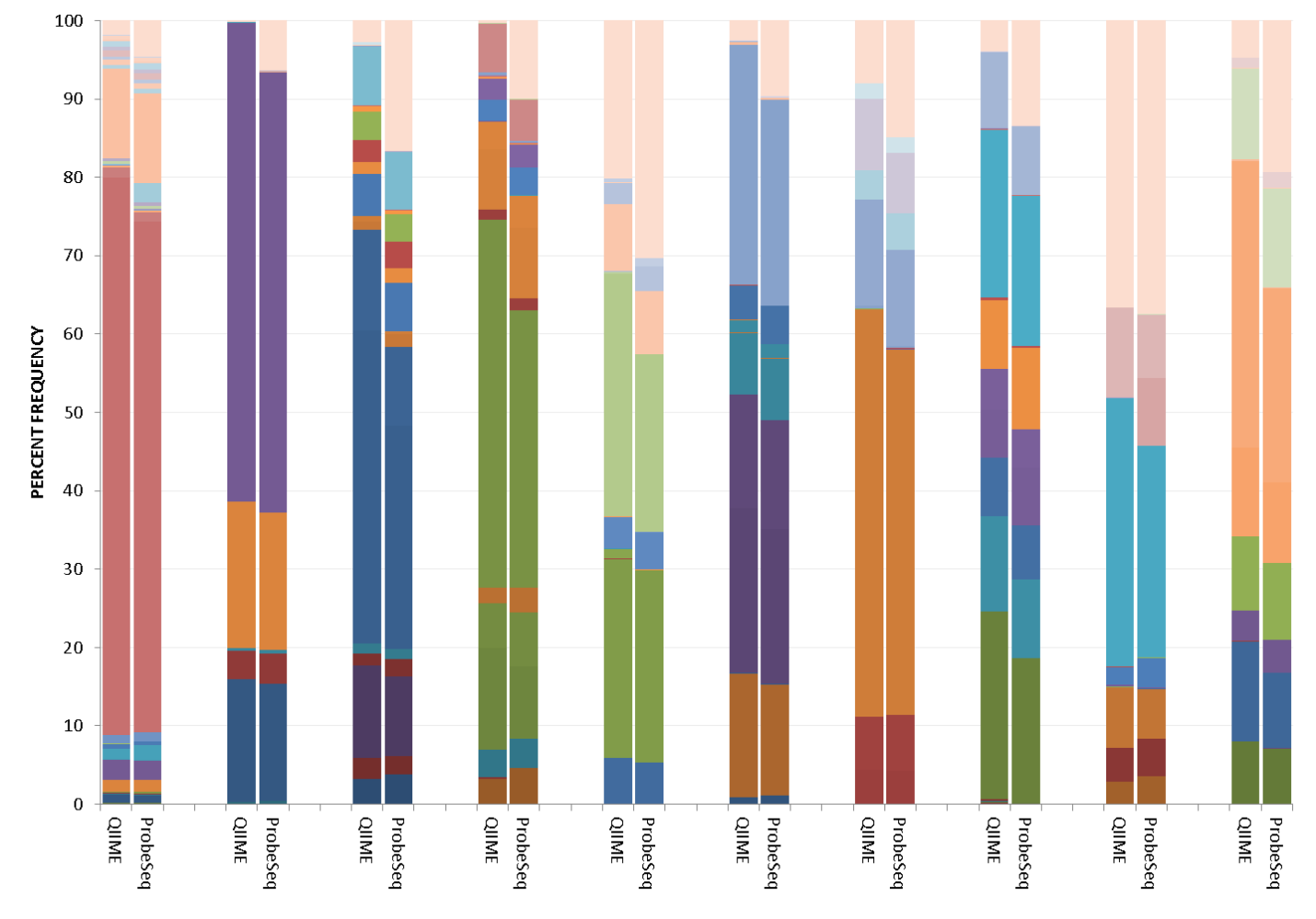
Probes were validated against 16S rRNA gene sequences in several databases including HOMD, GreenGenes, GenBank and the Ribosomal Database Project. In addition, sequences were validated in silico against several large datasets from our own studies (Dewhirst et al. 2010) in which there were typically multiple to hundreds of representatives for each taxon.